Iron's oxidation state in oxyhemoglobin
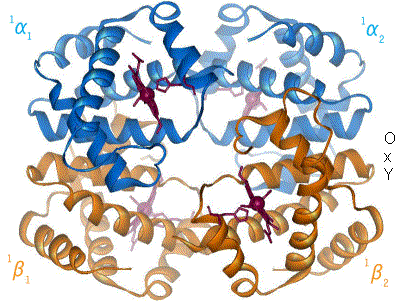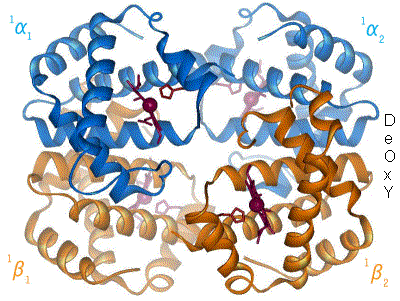
Assigning oxygenated hemoglobin's oxidation state is difficult because oxyhemoglobin (Hb-O2), by experimental measurement, is diamagnetic (no net unpaired electrons), yet the lowest-energy (ground-state) electron configurations in both oxygen and iron are paramagnetic (suggesting at least one unpaired electron in the complex). The lowest-energy form of oxygen, and the lowest energy forms of the relevant oxidation states of iron, are these:
- Triplet oxygen, the lowest-energy molecular oxygen species, has two unpaired electrons in antibonding π* molecular orbitals.
- Iron(II) tends to exist in a high-spin 3d6 configuration with four unpaired electrons.
- Iron(III) (3d5) has an odd number of electrons, and thus must have one or more unpaired electrons, in any energy state.
All of these structures are paramagnetic (have unpaired electrons), not diamagnetic. Thus, a non-intuitive (e.g., a higher-energy for at least one species) distribution of electrons in the combination of iron and oxygen must exist, in order to explain the observed diamagnetism and no unpaired electrons.
The two logical possibilities to produce diamagnetic (no net spin) Hb-O2 are:
- Low-spin Fe2+ binds to singlet oxygen. Both low-spin iron and singlet oxygen are diamagnetic. However, the single form of oxygen is the higher-energy form of the molecule.
- Low-spin Fe3+ binds to O2•− (the superoxide ion) and the two unpaired electrons couple antiferromagnetically, giving observed diamagnetic properties. Here, the iron has been oxidized (has lost one electron), and the oxygen has been reduced (has gained one electron).
Another possible model in which low-spin Fe4+ binds to peroxide, O22−, can be ruled out by itself, because the iron is paramagnetic (although the peroxide ion is diamagnetic). Here, the iron has been oxidized by two electrons, and the oxygen reduced by two electrons.
Direct experimental data:
- X-ray photoelectron spectroscopy suggests iron has an oxidation state of approximately 3.2.
- Infrared vibrational frequencies of the O-O bond suggests a bond length fitting with superoxide (a bond order of about 1.6, with superoxide being 1.5).
- X-ray Absorption Near Edge Structures at the iron K-edge. The energy shift of 5 eV between deoxyhemoglobin and oxyhemoglobin, as for all the methemoglobin species, strongly suggests an actual local charge closer to Fe3+ than Fe2+.
Thus, the nearest formal oxidation state of iron in Hb-O2 is the +3 state, with oxygen in the −1 state (as superoxide .O2−). The diamagnetism in this configuration arises from the single unpaired electron on superoxide aligning antiferromagnetically with the single unpaired electron on iron (in a low-spin d5 state), to give no net spin to the entire configuration, in accordance with diamagnetic oxyhemoglobin from experiment.
The second choice of the logical possibilities above for diamagnetic oxyhemoglobin being found correct by experiment, is not surprising: singlet oxygen (possibility #1) is an unrealistically high energy state. Model 3 leads to unfavorable separation of charge (and does not agree with the magnetic data), although it could make a minor contribution as a resonance form. Iron's shift to a higher oxidation state in Hb-O2 decreases the atom's size, and allows it into the plane of the porphyrin ring, pulling on the coordinated histidine residue and initiating the allosteric changes seen in the globulins.
Early postulates by bio-inorganic chemists claimed that possibility #1 (above) was correct and that iron should exist in oxidation state II. This conclusion seemed likely, since the iron oxidation state III as methemoglobin, when not accompanied by superoxide .O2− to "hold" the oxidation electron, was known to render hemoglobin incapable of binding normal triplet O2 as it occurs in the air. It was thus assumed that iron remained as Fe(II) when oxygen gas was bound in the lungs. The iron chemistry in this previous classical model was elegant, but the required presence of the diamagnetic, high-energy, singlet oxygen molecule was never explained. It was classically argued that the binding of an oxygen molecule placed high-spin iron(II) in an octahedral field of strong-field ligands; this change in field would increase the crystal field splitting energy, causing iron's electrons to pair into the low-spin configuration, which would be diamagnetic in Fe(II). This forced low-spin pairing is indeed thought to happen in iron when oxygen binds, but is not enough to explain iron's change in size. Extraction of an additional electron from iron by oxygen is required to explain both iron's smaller size and observed increased oxidation state, and oxygen's weaker bond.
The assignment of a whole-number oxidation state is a formalism, as the covalent bonds are not required to have perfect bond orders involving whole electron transfer. Thus, all three models for paramagnetic Hb-O2 may contribute to some small degree (by resonance) to the actual electronic configuration of Hb-O2. However, the model of iron in Hb-O2 being Fe(III) is more correct than the classical idea that it remains Fe(II).


Comments
Post a Comment